Brazil Tightens the Screws on Technology Transfer Pricing in Productive Development Partnerships: What the Ministry of Health's New Methodology Means for IP Holders

For more than a decade, Brazil's Productive Development Partnerships (PDPs) have served as one of the country's most strategically important industrial health-policy tools, channeling private-sector technology and manufacturing capability into the public sector to expand access to strategic medicines, vaccines, biologics, and medical devices through the Sistema Único de Saúde (SUS). For multinational pharmaceutical and biotechnology companies, PDPs are also one of the most significant, and procedurally distinctive, vehicles through which know-how, regulatory dossiers, and patent rights are transferred to Brazilian public laboratories.
On April 7, 2026, Brazil's Ministry of Health (MoH), through the Department of the Health Economic-Industrial Complex (DECIS) within the Secretariat of Science, Technology, and Innovation in Health (SCTIE), issued Technical Note No. 14/2026-DECIS/SCTIE/MS (Note), consolidating a structured methodology for valuing technology transfer in PDPs. The Note responds to long-standing determinations of Brazil's Federal Court of Accounts (Tribunal de Contas da União, or TCU), most recently in Panel Decision No. 1.014/2025, and follows the substantive pricing framework first introduced by Informative Note No. 17/2025-DECIS/SCTIE/MS in December 2025.
For practitioners advising clients on Brazilian PDP transactions, this is a meaningful development. The Note formalizes how the value of technology transfer, including know-how, technical and regulatory documentation, dossier rights, and IP transfers or licenses, is to be decomposed, audited, and, where necessary, reimbursed. It also signals a pending complementary regulation, expected by July 2026, that will embed these criteria into the formal regulatory architecture of PDPs under Ordinance GM/MS No. 4.472/2024. In this post, we walk through the new pricing model, highlight the IP-relevant components, and identify strategic considerations for foreign rights holders structuring or renegotiating PDP arrangements.
Background: PDPs and the Long Shadow of TCU Oversight
PDPs were designed as hybrid instruments that combine product supply with the gradual internalization of production and regulatory capacity by Brazilian public laboratories (Laboratórios Farmacêuticos Oficiais, or LFOs). A foreign or domestic private partner agrees to supply a strategic health product to the Ministry of Health while progressively transferring the technology, manufacturing know-how, analytical methods, regulatory dossiers, and, in many cases, IP rights or licenses, to a public partner over the life of the partnership. By the conclusion of the PDP, the public laboratory is intended to have full autonomy to manufacture, register, and supply the product.
The TCU, Brazil's federal audit authority, has scrutinized this model for years. In Panel Decision No. 725/2018, later upheld by Panel Decision No. 1.014/2025, the TCU determined that the Ministry of Health should “define the criteria and methodologies to be observed for calculating the value of technology transfer (know-how), including for purposes of establishing contractual penalties.” Notably, the TCU did not prescribe a single pricing formula. Instead, it required minimum guidelines capable of recognizing the diversity of PDP arrangements while ensuring transparency, traceability, and economic-financial balance.
Technical Note No. 14/2026 is the Ministry's most detailed response to date. It consolidates the work of Informative Note No. 17/2025, formally records its development and validation by the GECEIS (the inter-governmental executive group on the Health Economic-Industrial Complex, which approved the methodology unanimously on November 19, 2025), and adds operational details intended to support audit, contract negotiation, and indemnification calculations.
The Core Equation: Splitting Supply from Technology Transfer
At the heart of the Note is a foundational premise reaffirmed by the TCU: in a PDP, supply and technology transfer are inseparable. A PDP is neither a pure procurement contract nor a pure technology-transfer agreement. Removing either element changes the legal and economic nature of the arrangement. With that premise in mind, the Note decomposes the PDP price into two top-level components:
PDP Price = F + BDI
F (Fabricação/Fornecimento) — Manufacturing/Supply: the unit price for production and delivery of the product to the public partner, encompassing raw materials, packaging, direct labor, utilities, quality control, manufacturing overhead, supply logistics, manufacturing-related taxes, and the operational margin on supply.
BDI (Benefícios e Despesas Indiretas) — Indirect Benefits and Expenses: the consolidated rubric for the costs and charges associated with executing the PDP that are not directly attributable to a specific manufacturing input. This is where the Note introduces its most consequential framework.
BDI is decomposed into seven sub-components:
BDI = CI + T + MC + Ca + D + L + Ce
CI — Indirect Costs (central administration, uncertainty/guarantee margin, financial costs)
T — Tax Costs (PIS, COFINS, ISS, CPRB and other taxes on gross revenue)
MC — Contribution Margin for the public producer, intended to cover fixed costs and reinvestment
Ca — Training and Capacity Building (structured training plans, on-the-job training, technical missions, SOPs, train-the-trainer programs)
D — Regulatory Development (dossier compilation, ANVISA submissions, analytical and process validation, post-registration filings)
L — Technology Licensing (Phase III) — provisional licensing of the right to use the technology, often through a "clone" registration in the name of the LFO and/or co-titularity, allowing continuous supply after private-partner divestment and prior to definitive assignment
Ce — Valuation of Technology for Definitive Assignment — the integral, irrevocable transfer of the technology package, completed at the end of the PDP
Two further constraints frame the entire model. First, the total PDP price (including both F and BDI) must remain at or below the average price practiced within the SUS for the same product, calculated under Normative Instruction SEGES/ME No. 65/2021. Second, the TCU has reaffirmed that the parties cannot negotiate just the supply or just the technology transfer in isolation; the obligations are indissociable.
The IP-Relevant Components: L and Ce
From an intellectual property perspective, the most consequential elements of the new framework are the L (Licensing) and Ce (Definitive Assignment) components. These are the components in which IP-related assets are most directly concentrated, the temporary use and, ultimately, the irrevocable transfer of the technology package, including technical and regulatory documentation, dossiers, know-how, and IP rights or licenses where applicable.
The Note describes the Phase III Licensing component (L) as a precarious, time- and scope-limited right of use of the regulatory dossier and know-how during the final phase of the PDP. It is typically operationalized through a "clone" registration mechanism, that is, a marketing authorization in the name of the public laboratory, or co-titularity, that permits continuous supply once the private partner has begun its withdrawal but before the technology has been definitively assigned. Historically, the licensing of regulatory dossiers, including temporary clone registrations, has been a familiar arrangement among pharmaceutical companies in Brazil. The Note specifies that the licensing scope must address term, conditions of use, regulatory maintenance obligations, and performance milestones, ensuring traceability and governance.
The Definitive Assignment component (Ce) is the more substantive of the two. It corresponds to the integral and irrevocable transfer of the technology, with the private partner's complete withdrawal and full autonomy of the public producer. The Note specifies a minimum package that must be included:
Authorization to use the technical and industrial documentation associated with the dossier or clone registration, and its history;
Definitive assignment of technical, productive, and regulatory know-how, including specifications, methods, and critical parameters;
A commitment of non-opposition to future registrations, modifications, and expansions by the public producer;
A guarantee of full autonomy in production, registration, and post-registration filings; and
Transfer of intellectual property rights, or non-exclusive licenses, where applicable, with clearly defined scope and limitations.
That last bullet is one practitioners should pay close attention to. The Note explicitly contemplates two pathways for the IP component of Ce: a transfer of IP rights (assignment) or a non-exclusive license, with the choice depending on the nature of the underlying assets and the parties' commercial arrangement. For patent holders, this opens a meaningful degree of flexibility but also imposes discipline: scope and limitations must be clearly defined.
Ultimately, the key question will be whether the rights granted to the public laboratory are sufficient to allow it to manufacture, register, and commercialize the product autonomously after completion of the PDP. Vague or open-ended IP commitments embedded in PDP contracts will be far more difficult to defend under an audit-driven framework that rewards traceability and ex ante clarity.
Numerical Benchmarks: New Reference Ranges for Chemically Synthesized Medicines
One of the most concrete contributions of Technical Note No. 14/2026 is the introduction of numerical reference ranges for the BDI components. While Informative Note No. 17/2025 had identified L and Ce as part of the PDP pricing structure, the 2026 Note now goes further by identifying indicative percentage ranges for chemically synthesized medicines.
The Note is careful, however, not to present these ranges as universal benchmarks. It expressly states that the Ministry of Health does not yet have definitive parameters for acceptable percentage ranges for Technology Licensing – Phase III (L) and Definitive Assignment (Ce). Instead, it relies on selected market references for chemically synthesized medicines - including data from Micronomics LLP (which observed an average pharmaceutical royalty of 5.66%) and TT Consultants' analysis of European pharma royalty rates for products in late regulatory stages.
The Note draws on TCU Panel Decision No. 2.622/2013 (the TCU's foundational ruling on BDI methodology in public contracts) to set parameters for the more general BDI components, and on industry royalty data to set the IP-related ranges. The resulting reference parameters are summarized below:
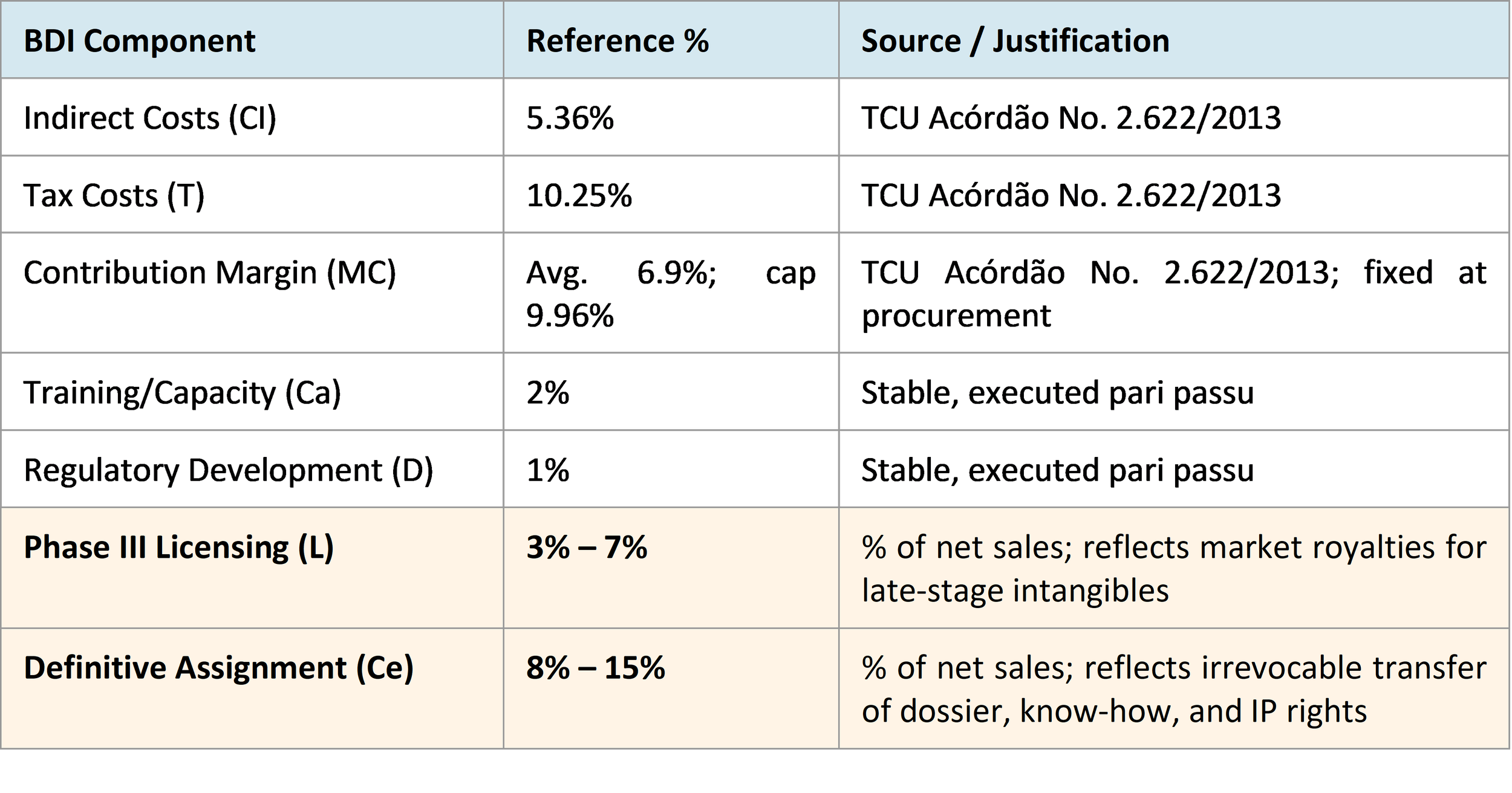
Importantly, the Note acknowledges that these ranges apply to chemically synthesized medicines, which are characterized by lower technological complexity, less reliance on tacit knowledge, and greater regulatory predictability. The Note expressly contemplates that biologics, vaccines, blood products, and medical devices may warrant different ranges, and that the Ministry has not yet established benchmarks for these higher-complexity categories. For practitioners working on biologic or biosimilar PDPs, this is a critical gap to monitor: the framework anticipates jurisdiction-specific differentiation, but the numerical benchmarks for biologics remain to be developed.
Fluctuating Versus Stable Components: A Negotiation Framework
The Note draws a sharp distinction between BDI components whose values may fluctuate over the life of a PDP and those that should remain stable, defined ex ante. This distinction matters because PDPs follow project-financing logic: their long-term economic-financial sustainability depends on cash-flow projections built on premises fixed at the start of the project. Allowing too many components to float undermines predictability and the original advantage analysis used to justify the partnership.
Fluctuating components: F (manufacturing/supply) may fluctuate with market price variations of the product. CI (indirect costs) and T (tax costs) vary proportionally with F.
Stable components:MC (contribution margin), Ca (training), and D (regulatory development) are fixed values that do not fluctuate. Notably, when MC is offered as a percentage by bidders during the public laboratory's selection process, it cannot be altered after selection, modification would violate the principle of isonomy in public procurement and compromise the integrity of the bid process.
Residual components:L (licensing) and Ce (definitive assignment) are calculated residually within the price formation and serve as the principal benchmark for reimbursement or indemnification calculations in the event of an unsuccessful technology transfer.
Reimbursement and Indemnification: The Ce Component as the Principal Concern
Perhaps the most important practical contribution of the Note, and one that practitioners advising private partners should examine carefully, is the framework for reimbursement in the event of early termination or failed technology transfer. The Note distinguishes among the components based on whether the corresponding consideration has already been earned through performance:
F (manufacturing) is remunerated per unit supplied; no reimbursement after supply has occurred.
CI and T are general, non-individualizable charges and are not reimbursable, consistent with established TCU jurisprudence.
MC is generally not refundable, though governance and monitoring clauses may permit partial or total recovery in cases of misappropriation or deviation of purpose.
Ca and D are executed gradually (pari passu) and are not reimbursable due to their immaterial and cumulative character.
L is treated as remuneration for the temporary, conditioned use of the technology during the PDP, and is not reimbursable once each supply cycle has been completed.
Ce is the principal locus of reimbursement risk. Because Ce is paid progressively through product purchases but corresponds to a transfer that is only completed at the end of the PDP, proportional reimbursement is appropriate if the partnership terminates early or if the technology transfer is not completed. Reimbursement is calculated proportionally based on what was already paid before interruption.
Independent of any reimbursement of technology-transfer costs, contractual penalties for non-performance, including default fines, administrative sanctions, and other legal remedies — remain applicable. Private partners should therefore expect parallel exposure: a proportional refund obligation on the Ce component plus contractual penalty provisions, both potentially triggered by the same termination event.
Reimbursement obligations in the PDP context are not a new issue. Under the previous PDP framework, several partnerships struggled to demonstrate that technology transfer had in fact been completed. In some cases, the issue escalated into litigation. Last year, an emblematic trial decision concerning a PDP between a generic company and a public institution partner to produce an oncology product set an important precedent on who may be held liable, and how liability should be quantified, when the technology transfer process is not fully concluded.
The plaintiff argued that technology transfer had been remunerated but not executed, seeking full reimbursement of all amounts invoiced under the PDP. The court, however, found that between 2013 and 2018 the medicines were effectively delivered to the Ministry of Health in adequate quantities and regularly supplied to SUS patients. At the same time, it concluded that technology transfer had not occurred, thereby frustrating the essential purpose that justified the differentiated contractual structure and the payment of prices above market levels.
In this context, the judgment indicates that the private partner may be held liable for damage to the public treasury even in the absence of a finding that it directly caused the interruption of the technology transfer process. The compensable harm corresponds to the economic value attributed to the technology-transfer obligation, i.e., the portion of PDP payments that cannot be justified by the mere supply of the product where the promised transfer, serving as the causal basis for the exceptional procurement arrangement, is not effectively delivered.
With respect to the allocation of responsibility, the decision established that even if the failure to conclude the transfer resulted from circumstances external to the private partner, knowledge of the impediments triggered a duty to act.
Accordingly, the private partner should have proposed concrete alternatives to enable contract performance or, if performance proved unfeasible, suspended execution and returned amounts unduly received. The judgment thus articulates a positive legal duty on the private partner not to allow the unjustified continuation of a contractual execution that frustrates technology transfer.
Recognizing the strong public interest underlying the contracting, namely, the regular and continuous supply of an essential oncological medicine to the SUS, the court held that full nullification of the contracts and related acts would be disproportionate and incompatible with legal certainty. Consequently, it determined that the amounts paid by the Union could be analytically decomposed into two legally distinct portions: (i) a first portion corresponding to the value of the medicines effectively supplied, which remunerates an obligation duly performed; and (ii) a second portion corresponding to the overprice paid in relation to market values, which exclusively remunerates the obligation to transfer technology.
However, because the value of the technology was not contractually discriminated, the court held that, absent clear and unequivocal documentary evidence of accounting separation between supply payments and technology-transfer remuneration, the liquidation phase must necessarily adopt an objective benchmark: the prices practiced in the first public bid conducted after the termination of the PDP (Public Bid No. 98/2018), whose prices were deemed to reflect market values under free competition, without embedded technology-transfer costs.
The case should be closely monitored as it progresses through pending motions for clarification and appellate review. More broadly, it illustrates why the new methodology should be read not merely as a pricing tool, but as a response to a concrete enforcement problem: how to identify, document, justify, monitor, and, where applicable, reimburse the economic value attributed to technology transfer. For industry, the message is that transparency will be expected throughout the entire lifecycle of the PDP - from price formation and contractual allocation of value to ongoing monitoring, documentation, and evidence of effective technology absorption by the public laboratory.
Strategic Implications for Foreign Patent Holders and IP Practitioners
Several aspects of the new framework warrant immediate attention from patent counsel and licensing executives advising clients with Brazilian PDP exposure.
First, the Ce component's IP package will require careful drafting. The Note expressly contemplates either a transfer of IP rights or a non-exclusive license, with scope and limitations "clearly defined." In our assessment, this is an opportunity for sophisticated drafting. Private partners that wish to retain commercial rights outside the SUS context should negotiate non-exclusive licenses with carefully delineated field-of-use limitations rather than outright assignments. The Note provides explicit textual support for this approach, which can reduce the IP cost of participating in a PDP for patent owners with global commercial strategies.
Second, the non-opposition commitment deserves close attention.The Ce package requires a commitment of non-opposition to future registrations, modifications, and expansions by the public producer. For patent holders, this could be construed broadly to include not just regulatory non-opposition but also restraints on enforcement of related patent rights. Counsel should negotiate the precise scope of non-opposition, specifically distinguishing regulatory non-opposition from any covenant not to assert patents, to avoid inadvertently creating broader IP forbearance than intended.
Third, the L percentage range (3–7%) and Ce range (8–15%) for chemically synthesized medicines are now anchored to market royalty data.Practitioners negotiating PDPs for higher-complexity products — biologics, biosimilars, vaccines, advanced therapies, should expect upward pressure when benchmarks are eventually developed for those categories, given the greater reliance on tacit knowledge and the higher commercial value of those technologies. Foreign rights holders should be prepared to substantiate higher percentages with documented market royalty data and complexity justifications.
Fourth, the residual nature of L and Ce in the price equation means that they bear the brunt of reimbursement risk.Private partners contributing significant IP to a PDP should carefully evaluate the proportional reimbursement obligations attached to Ce. Where the technology package includes patent rights or complex know-how that has been gradually transferred, the partner may face material refund exposure if the PDP is terminated before completion of the definitive assignment. Contracts should specify the proportional formula precisely, address what evidence will support the calculation, and consider whether milestones tied to technology-transfer completeness can offset the residual exposure.
Fifth, the clone-registration mechanism for L is a useful but underappreciated tool. The Note formalizes the long-standing Brazilian practice of using clone registrations and co-titularity to bridge the gap between private-partner divestment and definitive technology assignment. Patent counsel should ensure that clone registrations and any associated regulatory submissions are clearly within the scope of the licensed rights, with appropriate terms, performance milestones, and post-registration maintenance obligations specified, particularly given that the Note now requires this level of detail.
Sixth, the July 2026 complementary regulation will be a watershed moment.The Ministry has signaled that a binding regulation will be issued by July 2026 to incorporate this methodology, monitoring mechanisms, and governance improvements into the formal regulatory framework under Ordinance GM/MS No. 4.472/2024. When that happens, the percentage ranges, decomposition requirements, and reimbursement rules will move from methodological guidance to enforceable regulation, with potential implications for contract structuring, cost transparency, auditability, and the legal enforceability of indemnification mechanisms. Practitioners should plan to revisit any PDP contracts negotiated between now and mid-2026 once the binding regulation is issued, particularly in regard to ex ante parameter fixation and reimbursement clauses.
A Note on Global Perspective
Brazil's approach is notable in the broader landscape of public-private technology-transfer frameworks. Few jurisdictions have moved as decisively to formalize the decomposition of supply price from technology-transfer value, and fewer still have set numerical reference ranges for the IP-licensing and definitive-assignment components. India's compulsory licensing regime, China's evolving IP framework for state-procurement scenarios, and South Africa's local-content requirements each address aspects of the same broader policy concern, how to balance access to strategic health technologies with fair compensation for IP holders, but none has produced a methodology of comparable granularity. For multinational rights holders, Brazil's framework may serve as a reference point in negotiations elsewhere, both because it provides quantitative benchmarks and because its emphasis on transparency and traceability is increasingly aligned with what audit-conscious procurement authorities are seeking globally.
Key Takeaways
Brazil's Ministry of Health has consolidated a structured methodology for valuing technology transfer in PDPs, decomposing the PDP price into manufacturing (F) and indirect benefits and expenses (BDI), with BDI further decomposed into seven sub-components.
Reference percentage ranges now apply to chemically synthesized medicines: Phase III Licensing (L) at 3–7% of net sales and Definitive Assignment (Ce) at 8–15% of net sales. Benchmarks for biologics, vaccines, and medical devices have not yet been established and will be a critical area to monitor.
The Ce component is the principal locus of IP transfer and reimbursement risk. It contemplates either a transfer of IP rights or a non-exclusive license, with clearly defined scope and limitations. Private partners face proportional reimbursement obligations if the PDP is terminated before completion of definitive assignment.
The non-opposition commitment within Ce should be carefully scoped to distinguish regulatory non-opposition from any broader covenant not to assert patent rights — the difference can be material to a multinational's global IP strategy.
Phase III Licensing (L) is operationalized through clone-registration mechanisms or co-titularity, with the Note now requiring detailed specification of term, conditions of use, regulatory maintenance obligations, and performance milestones.
A binding complementary regulation is expected by July 2026 to incorporate the methodology into Ordinance GM/MS No. 4.472/2024. Practitioners should plan to revisit PDP contracts negotiated between now and mid-2026 in light of the forthcoming regulation, particularly regarding reimbursement and indemnification clauses.
The MC, Ca, and D components are fixed ex ante and cannot fluctuate. Where MC is bid as a percentage during the public laboratory's selection of a private partner, it cannot be altered post-selection without violating Brazilian public procurement law.
We recommend that practitioners review all active and pending PDP arrangements for clients with Brazilian exposure to (i) confirm that L and Ce components are clearly delineated; (ii) ensure that IP transfer or licensing scope is precisely defined; (iii) evaluate proportional reimbursement exposure under the Ce component; and (iv) prepare for renegotiation as the July 2026 binding regulation takes shape.
This post was written by Lisa Mueller, Rob Rodriques and Dara Offrede of RNA Law.